Understanding the complexity of cancer requires methods that can integrate equally complex biological data. In our lab, we are committed to developing probabilistic models that bring together multiple molecular layers, including genomics, epigenomics, transcriptomics, proteomics and metabolomics, to provide a holistic view of each patient. These models uncover hidden structure by capturing both shared and modality-specific variation, allowing us to reduce noise and reveal biologically meaningful patterns. By modeling system-level responses to perturbations such as drug treatments or environmental changes, we aim to generate representations that are not only statistically robust but also interpretable, enabling new biological insights that can be directly validated and translated into clinical understanding.
MuVI
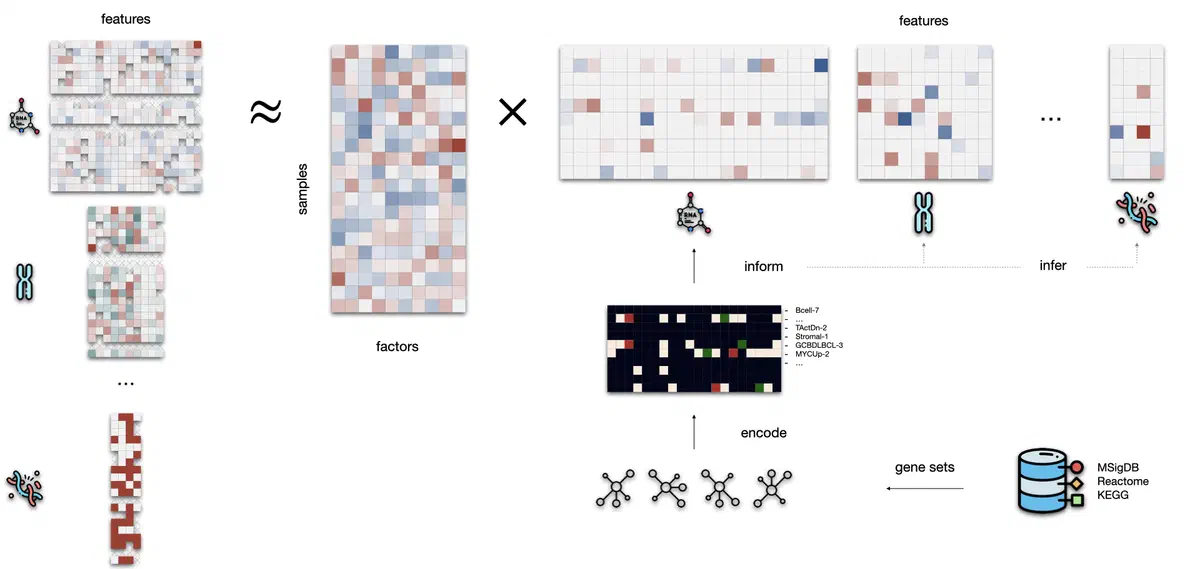
MuVI is a general-purpose probabilistic latent variable model for multi-omics integration that incorporates prior biological knowledge into its structure. It uses pathway annotations, gene sets, or cell-type signatures to guide the discovery of latent factors that explain variation across different data types. Even when this prior knowledge is noisy or incomplete, MuVI is able to learn biologically relevant dimensions, enabling scientists to interpret the sources of variation in the data more clearly and to relate them to known mechanisms.
MOMO-GP (Multi-Omic Multi-output Gaussian Processes) addresses the challenge of learning interpretable representations from single-cell multi-omics data, which are typically high-dimensional, sparse, and nonlinear. Unlike traditional methods that trade off interpretability for modeling power, MOMO-GP combines neural networks with Gaussian Processes to achieve both. It learns separate latent embeddings for cells and features, as well as shared and modality-specific components in the multi-view setting. By modeling gene relevance explicitly, MOMO-GP connects cell clusters to marker genes, making the learned structure readily interpretable in biological terms.
JOANA is a probabilistic model for pathway enrichment analysis (PEA) that overcomes limitations of classical approaches like Over-Representation Analysis (ORA) and Functional Class Scoring (FCS). While methods such as GSEA work with continuous scores, they typically operate on a single omics layer and can yield overly broad sets of enriched pathways. JOANA improves on this by modeling enrichment scores across multiple omics layers using mixtures of beta distributions within a Bayesian framework. This allows it to estimate the probability of pathway enrichment both within and across modalities, yielding higher precision and more biologically relevant results.
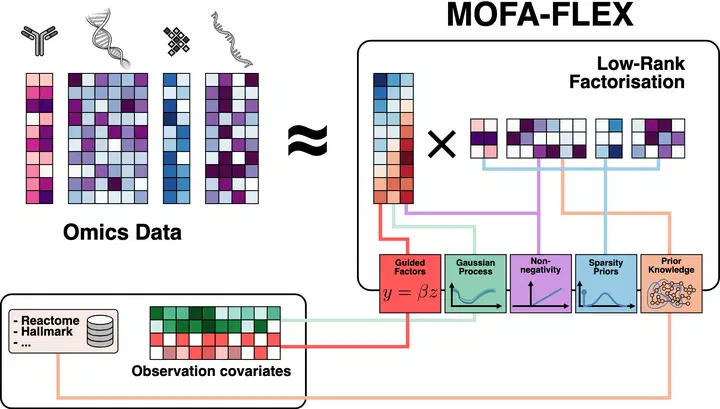
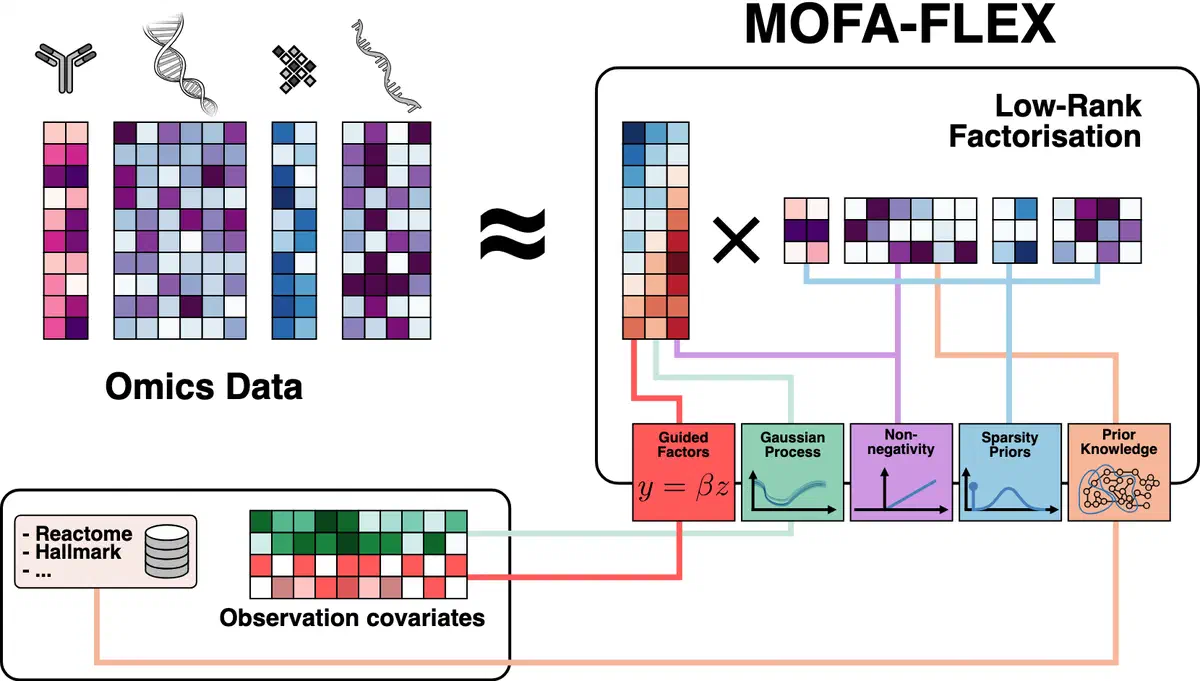
MOFA-FLEX is our upcoming framework for flexible and interpretable multi-omics integration. Designed to generalize the principles behind models like MuVI and MUSIC, MOFA-FLEX supports heterogeneous data types, modular priors, and scalable inference. Its architecture allows for tailored modeling of real-world datasets, balancing interpretability with modeling flexibility. MOFA-FLEX is currently under active development and will provide a unified foundation for future applications in cancer biology and beyond.
PACMON is a scalable Bayesian factor model for interpreting high-throughput single-cell perturbation screens. It uses pathway priors to infer molecular programs and quantify how genetic or chemical perturbations affect these programs across modalities. Applied to multimodal Perturb-CITE-seq data and atlas-sized chemical perturbation screens, PACMON identifies coherent immune-, cell-state-, and drug-response programs, linking perturbations to pathway-level molecular changes in an interpretable and scalable way.
MANTRA (Multi-view ANalysis with Tensor and matRix Alignment) is a Bayesian framework for integrating matrices and higher-order tensors in multi-omics studies. It preserves experimental structures such as patient × drug × dose tensors while jointly modeling them with standard omics matrices. By learning sparse, interpretable latent factors and handling missing data naturally, MANTRA uncovers clinically relevant patient subgroups and cell-type-specific disease programs that can be missed by conventional matrix-based integration methods.